Intrinsic-reaction-coordinate calculations: Difference between revisions
| Line 65: | Line 65: | ||
The INCAR file located in directory p should differ from that in directory m only in the value of parameter IRC_DIRECTION, which should be set to positive 1. | The INCAR file located in directory p should differ from that in directory m only in the value of parameter IRC_DIRECTION, which should be set to positive 1. | ||
[[Media:IrcExample1.png]] | |||
Both calculations can be finished in just a few seconds and the potential energy profile along IRC: | Both calculations can be finished in just a few seconds and the potential energy profile along IRC: | ||
| Line 79: | Line 79: | ||
sort -n tmp.tmp >ircShift.dat | sort -n tmp.tmp >ircShift.dat | ||
executed in a directory one level above directories m and p generates the file ircShift.dat containing data in two-column format that can be visualized using gnuplot, xmgrace, origin, or many other popular visualization tools. | executed in a directory one level above directories m and p generates the file ircShift.dat containing data in two-column format that can be visualized using gnuplot, xmgrace, origin, or many other popular visualization tools. | ||
== Related tags and articles == | == Related tags and articles == | ||
Revision as of 16:04, 30 November 2023
The potential energy profiles along the intrinsic reaction coordinate (IRC) can be computed via the method of Hratchian and Schlegel[1]. The algorithm starts from the transition state and propagates the system via the damped-velocity-Verlet algorithm. The damping is realized via rescaling the velocity vector to a constant value () after each propagation step. At the same time, the time step is adaptively changed so as to ensure that the trajectory generated by the algorithm does not differ from true IRC by more than the predefined tolerance factor . As an input, the structure of a well-relaxed transition state and the direction of the unstable vibration mode must be provided. For that purpose, a CONTCAR file from an improved-dimer-method calculation converged with a tight relaxation criterion (e.g., EDIFFG =-0.005) can be used. To obtain a complete energy profile along the IRC connecting two stable states, two independent calculations with positive (IRC_DIRECTION =1) and negative (IRC_DIRECTION =-1) initial displacement along the direction of the unstable mode must be performed.
The following parameters can be modified to affect the performance of the method:
- IRC_DIRECTION direction of the initial displacement (-1|1 – negative|positive)
- IRC_STOP the number of steps the energy must monotonously increase before the algorithm terminates. In order to avoid a premature termination, especially close to transition states., e.g., due to numerical noise, IRC_STOP should always be greater than 1.
- IRC_DELTA0 the tolerance factor in Å
- IRC_MINSTEP specifies the lower limit for the time step in fs
- IRC_MAXSTEP specifies the upper limit for the time step in fs
- IRC_VNORM0 the value of in Å/fs - the smaller the value, the closer the computed trajectory follows the true IRC (but the more ionic steps are required, which might be a limitation if the calculation is performed at a DFT level)
| Mind: This method is presently available only for fixed cell shape (i.e., ISIF = 2) simulations. |
| Mind: The calculation must be initialized from a very well-relaxed transition state (EDIFFG = -0.005 or less in absolute value). |
| Mind: This type of calculation can be also performed at the MLFF level. |
Practical example
As practical example, let us consider the SN2 reaction of CH3Cl with Cl- for which we wish to determine potential energy profile along IRC using machine learned forcefield trained to reproduce PBE density functional approximation. The IRC calculation consists of two independent simulations corresponding to parts of path linking transition state with reactant and with product, respectively. For practical reasons, we shall therefore run the simulations in two different directories called, say, m and p, both of which should contain POSCAR, KPOINTS, POTCAR, INCAR and ML_FF files. The POSCAR file:
transition state for the SN2 reaction of CH3Cl with Cl-
1.00000000000000
12.0000000000000000 0.0000000000000000 0.0000000000000000
0.0000000000000000 12.0000000000000000 0.0000000000000000
0.0000000000000000 0.0000000000000000 12.0000000000000000
C H Cl
1 3 2
Direct
0.5850734246784209 0.5520914206000113 0.6939676081570985 ! coordinates for atom 1
0.6198799799863561 0.5166828822917992 0.6192173893914401
0.5120438703668218 0.5155341457726120 0.7311383922253840
0.6232988535554732 0.6240585575728448 0.7315296645899942
0.4727762112937258 0.6675904758190063 0.5870243322899322
0.6973145053782144 0.4366510550400121 0.8010186348593505 ! coordinates for atom N
! unstable direction optimized by the dimer method
0.50309310E+00 -0.52116977E+00 0.48103627E+00 ! components for atom 1
-0.29990134E-01 0.31072124E-01 -0.24160721E-01
-0.37734828E-01 0.43431417E-01 -0.39422281E-01
-0.41647034E-01 0.41752749E-01 -0.38921427E-01
-0.20205452E+00 0.20762993E+00 -0.17619553E+00
-0.19166655E+00 0.19728353E+00 -0.20233630E+00 ! components for atom N
is simply a copy of a CONTCAR file from a well converged improved dimer calculation. Notice that besides usual structural input, POSCAR must contain unstable direction that is needed to initialize the algorithm. To obtain the ML_FF used in this example, the data included in compressed directory File:GenerateMLFF.zip should be used. The POTCAR file should be prepared for atoms C, H, and Cl and the KPOINTS file
Automatic 0 Gamma 1 1 1 0. 0. 0.
The INCAR file located in directory m should contain the following lines:
IBRION = 40 # invokes the IRC calculation
IRC_DIRECTION = -1 # negative initial direction of movement
IRC_STOP = 20 # terminate when IRC_STOP energies in row increase
IRC_VNORM0 = 0.0005 # affects accuracy, the smaller the better but the more
# ionic steps needed
NSW = 5000 # maximal number of steps
ISIF = 2
NELECT = 22 # the overall charge of this
# system is -1 hence the number
# of electrons is increased (not really needed when
# the calculation is done at the MLFF level)
ML_LMLFF = .TRUE. # invoke MLFF
ML_ISTART = 2 # sets the mode of MLFF to production
The INCAR file located in directory p should differ from that in directory m only in the value of parameter IRC_DIRECTION, which should be set to positive 1.
{kind=link}
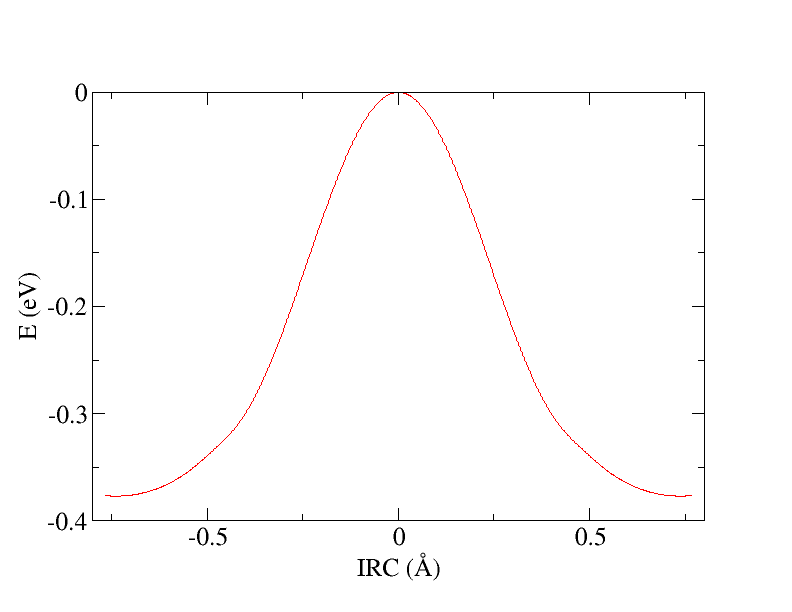
Both calculations can be finished in just a few seconds and the potential energy profile along IRC:
shown on the figure below, can be obtained by a simple post-processing. For instance, this simple bash script ircShift.sh:
#!/usr/bin/bash
e0=$(grep "IRC (A):" m/OUTCAR|awk '{print $5}'|head -1)
grep "IRC (A):" m/OUTCAR|awk '{print $3,$5 - "'${e0}'"}' >tmp.tmp
grep "IRC (A):" p/OUTCAR|awk '{print $3,$5 - "'${e0}'"}' >>tmp.tmp
sort -n tmp.tmp >ircShift.dat
executed in a directory one level above directories m and p generates the file ircShift.dat containing data in two-column format that can be visualized using gnuplot, xmgrace, origin, or many other popular visualization tools.
Related tags and articles
IRC_DIRECTION, IRC_STOP, IRC_DELTA0, IRC_MINSTEP, IRC_MAXSTEP, IRC_VNORM0